Rutherfordium
| Rutherfordium | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pronunciation |
|
||||||||||||||||||||||||||||||||||
| Mass number | [267] | ||||||||||||||||||||||||||||||||||
| Rutherfordium in the periodic table | |||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 104 | ||||||||||||||||||||||||||||||||||
| Group | group 4 | ||||||||||||||||||||||||||||||||||
| Period | period 7 | ||||||||||||||||||||||||||||||||||
| Block | d-block | ||||||||||||||||||||||||||||||||||
| Electron configuration | [Rn] 5f14 6d2 7s2 | ||||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 8, 18, 32, 32, 10, 2 | ||||||||||||||||||||||||||||||||||
| Physical properties | |||||||||||||||||||||||||||||||||||
| Phase at STP | solid (predicted) | ||||||||||||||||||||||||||||||||||
| Melting point | 2400 K (2100 °C, 3800 °F) (predicted) | ||||||||||||||||||||||||||||||||||
| Boiling point | 5800 K (5500 °C, 9900 °F) (predicted) | ||||||||||||||||||||||||||||||||||
| Density (near r.t.) | 17 g/cm3 (predicted) | ||||||||||||||||||||||||||||||||||
| Atomic properties | |||||||||||||||||||||||||||||||||||
| Oxidation states | (+2), (+3), +4 (parenthesized: prediction) | ||||||||||||||||||||||||||||||||||
| Ionization energies |
|
||||||||||||||||||||||||||||||||||
| Atomic radius | empirical: 150 pm (estimated) | ||||||||||||||||||||||||||||||||||
| Covalent radius | 157 pm (estimated) | ||||||||||||||||||||||||||||||||||
| Other properties | |||||||||||||||||||||||||||||||||||
| Natural occurrence | synthetic | ||||||||||||||||||||||||||||||||||
| Crystal structure | hexagonal close-packed (hcp) (predicted) |
||||||||||||||||||||||||||||||||||
| CAS Number | 53850-36-5 | ||||||||||||||||||||||||||||||||||
| History | |||||||||||||||||||||||||||||||||||
| Naming | after Ernest Rutherford | ||||||||||||||||||||||||||||||||||
| Discovery | Joint Institute for Nuclear Research and Lawrence Berkeley National Laboratory (1964, 1969) | ||||||||||||||||||||||||||||||||||
| Isotopes of rutherfordium | |||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||
Rutherfordium is a synthetic chemical element; it has symbol Rf and atomic number 104. It is named after physicist Ernest Rutherford. As a synthetic element, it is not found in nature and can only be made in a particle accelerator. It is radioactive; the most stable known isotope, 267Rf, has a half-life of about 48 minutes.
In the periodic table, it is a d-block element and the second of the fourth-row transition elements. It is in period 7 and is a group 4 element. Chemistry experiments have confirmed that rutherfordium behaves as the heavier homolog to hafnium in group 4. The chemical properties of rutherfordium are characterized only partly. They compare well with the other group 4 elements, even though some calculations had indicated that the element might show significantly different properties due to relativistic effects.
In the 1960s, small amounts of rutherfordium were produced at Joint Institute for Nuclear Research in the Soviet Union and at Lawrence Berkeley National Laboratory in California. Priority of discovery and hence the name of the element was disputed between Soviet and American scientists, and it was not until 1997 that the International Union of Pure and Applied Chemistry (IUPAC) established rutherfordium as the official name of the element.
Introduction
Synthesis of superheavy nuclei
A superheavy atomic nucleus is created in a nuclear reaction that combines two other nuclei of unequal size into one; roughly, the more unequal the two nuclei in terms of mass, the greater the possibility that the two react. The material made of the heavier nuclei is made into a target, which is then bombarded by the beam of lighter nuclei. Two nuclei can only fuse into one if they approach each other closely enough; normally, nuclei (all positively charged) repel each other due to electrostatic repulsion. The strong interaction can overcome this repulsion but only within a very short distance from a nucleus; beam nuclei are thus greatly accelerated in order to make such repulsion insignificant compared to the velocity of the beam nucleus. The energy applied to the beam nuclei to accelerate them can cause them to reach speeds as high as one-tenth of the speed of light. However, if too much energy is applied, the beam nucleus can fall apart.
Coming close enough alone is not enough for two nuclei to fuse: when two nuclei approach each other, they usually remain together for approximately 10−20 seconds and then part ways (not necessarily in the same composition as before the reaction) rather than form a single nucleus. This happens because during the attempted formation of a single nucleus, electrostatic repulsion tears apart the nucleus that is being formed. Each pair of a target and a beam is characterized by its cross section—the probability that fusion will occur if two nuclei approach one another expressed in terms of the transverse area that the incident particle must hit in order for the fusion to occur. This fusion may occur as a result of the quantum effect in which nuclei can tunnel through electrostatic repulsion. If the two nuclei can stay close for past that phase, multiple nuclear interactions result in redistribution of energy and an energy equilibrium.
| External videos | |
|---|---|
|
|
The resulting merger is an excited state—termed a compound nucleus—and thus it is very unstable. To reach a more stable state, the temporary merger may fission without formation of a more stable nucleus. Alternatively, the compound nucleus may eject a few neutrons, which would carry away the excitation energy; if the latter is not sufficient for a neutron expulsion, the merger would produce a gamma ray. This happens in approximately 10−16 seconds after the initial nuclear collision and results in creation of a more stable nucleus. The definition by the IUPAC/IUPAP Joint Working Party (JWP) states that a chemical element can only be recognized as discovered if a nucleus of it has not decayed within 10−14 seconds. This value was chosen as an estimate of how long it takes a nucleus to acquire its outer electrons and thus display its chemical properties.
Decay and detection
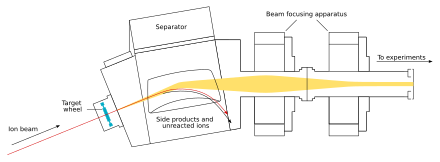
The beam passes through the target and reaches the next chamber, the separator; if a new nucleus is produced, it is carried with this beam. In the separator, the newly produced nucleus is separated from other nuclides (that of the original beam and any other reaction products) and transferred to a surface-barrier detector, which stops the nucleus. The exact location of the upcoming impact on the detector is marked; also marked are its energy and the time of the arrival. The transfer takes about 10−6 seconds; in order to be detected, the nucleus must survive this long. The nucleus is recorded again once its decay is registered, and the location, the energy, and the time of the decay are measured.
Stability of a nucleus is provided by the strong interaction. However, its range is very short; as nuclei become larger, its influence on the outermost nucleons (protons and neutrons) weakens. At the same time, the nucleus is torn apart by electrostatic repulsion between protons, and its range is not limited. Total binding energy provided by the strong interaction increases linearly with the number of nucleons, whereas electrostatic repulsion increases with the square of the atomic number, i.e. the latter grows faster and becomes increasingly important for heavy and superheavy nuclei. Superheavy nuclei are thus theoretically predicted and have so far been observed to predominantly decay via decay modes that are caused by such repulsion: alpha decay and spontaneous fission. Almost all alpha emitters have over 210 nucleons, and the lightest nuclide primarily undergoing spontaneous fission has 238. In both decay modes, nuclei are inhibited from decaying by corresponding energy barriers for each mode, but they can be tunnelled through.
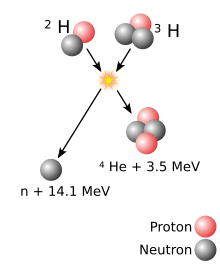
Alpha particles are commonly produced in radioactive decays because mass of an alpha particle per nucleon is small enough to leave some energy for the alpha particle to be used as kinetic energy to leave the nucleus. Spontaneous fission is caused by electrostatic repulsion tearing the nucleus apart and produces various nuclei in different instances of identical nuclei fissioning. As the atomic number increases, spontaneous fission rapidly becomes more important: spontaneous fission partial half-lives decrease by 23 orders of magnitude from uranium (element 92) to nobelium (element 102), and by 30 orders of magnitude from thorium (element 90) to fermium (element 100). The earlier liquid drop model thus suggested that spontaneous fission would occur nearly instantly due to disappearance of the fission barrier for nuclei with about 280 nucleons. The later nuclear shell model suggested that nuclei with about 300 nucleons would form an island of stability in which nuclei will be more resistant to spontaneous fission and will primarily undergo alpha decay with longer half-lives. Subsequent discoveries suggested that the predicted island might be further than originally anticipated; they also showed that nuclei intermediate between the long-lived actinides and the predicted island are deformed, and gain additional stability from shell effects. Experiments on lighter superheavy nuclei, as well as those closer to the expected island, have shown greater than previously anticipated stability against spontaneous fission, showing the importance of shell effects on nuclei.
Alpha decays are registered by the emitted alpha particles, and the decay products are easy to determine before the actual decay; if such a decay or a series of consecutive decays produces a known nucleus, the original product of a reaction can be easily determined. (That all decays within a decay chain were indeed related to each other is established by the location of these decays, which must be in the same place.) The known nucleus can be recognized by the specific characteristics of decay it undergoes such as decay energy (or more specifically, the kinetic energy of the emitted particle). Spontaneous fission, however, produces various nuclei as products, so the original nuclide cannot be determined from its daughters.
The information available to physicists aiming to synthesize a superheavy element is thus the information collected at the detectors: location, energy, and time of arrival of a particle to the detector, and those of its decay. The physicists analyze this data and seek to conclude that it was indeed caused by a new element and could not have been caused by a different nuclide than the one claimed. Often, provided data is insufficient for a conclusion that a new element was definitely created and there is no other explanation for the observed effects; errors in interpreting data have been made.History
Discovery
Rutherfordium was reportedly first detected in 1964 at the Joint Institute for Nuclear Research at Dubna (Soviet Union at the time). Researchers there bombarded a plutonium-242 target with neon-22 ions and separated the reaction products by gradient thermochromatography after conversion to chlorides by interaction with ZrCl4. The team identified spontaneous fission activity contained within a volatile chloride portraying eka-hafnium properties. Though a half-life was not accurately determined, later calculations indicated that the product was most likely rutherfordium-259 (259Rf in standard notation):
-
242
94Pu + 22
10Ne → 264−x
104Rf → 264−x
104RfCl4
In 1969, researchers at University of California, Berkeley conclusively synthesized the element by bombarding a californium-249 target with carbon-12 ions and measured the alpha decay of 257Rf, correlated with the daughter decay of nobelium-253:
-
249
98Cf + 12
6C → 257
104Rf + 4
n
The American synthesis was independently confirmed in 1973 and secured the identification of rutherfordium as the parent by the observation of K-alpha X-rays in the elemental signature of the 257Rf decay product, nobelium-253.
Naming controversy


As a consequence of the initial competing claims of discovery, an element naming controversy arose. Since the Soviets claimed to have first detected the new element they suggested the name kurchatovium (Ku) in honor of Igor Kurchatov (1903–1960), former head of Soviet nuclear research. This name had been used in books of the Soviet Bloc as the official name of the element. The Americans, however, proposed rutherfordium (Rf) for the new element to honor New Zealand physicist Ernest Rutherford, who is known as the "father" of nuclear physics. In 1992, the IUPAC/IUPAP Transfermium Working Group (TWG) assessed the claims of discovery and concluded that both teams provided contemporaneous evidence to the synthesis of element 104 and that credit should be shared between the two groups.
The American group wrote a scathing response to the findings of the TWG, stating that they had given too much emphasis on the results from the Dubna group. In particular they pointed out that the Russian group had altered the details of their claims several times over a period of 20 years, a fact that the Russian team does not deny. They also stressed that the TWG had given too much credence to the chemistry experiments performed by the Russians and accused the TWG of not having appropriately qualified personnel on the committee. The TWG responded by saying that this was not the case and having assessed each point raised by the American group said that they found no reason to alter their conclusion regarding priority of discovery. The IUPAC finally used the name suggested by the American team (rutherfordium).
The International Union of Pure and Applied Chemistry (IUPAC) adopted unnilquadium (Unq) as a temporary, systematic element name, derived from the Latin names for digits 1, 0, and 4. In 1994, IUPAC suggested a set of names for elements 104 through 109, in which dubnium (Db) became element 104 and rutherfordium became element 106. This recommendation was criticized by the American scientists for several reasons. Firstly, their suggestions were scrambled: the names rutherfordium and hahnium, originally suggested by Berkeley for elements 104 and 105, were respectively reassigned to elements 106 and 108. Secondly, elements 104 and 105 were given names favored by JINR, despite earlier recognition of LBL as an equal co-discoverer for both of them. Thirdly and most importantly, IUPAC rejected the name seaborgium for element 106, having just approved a rule that an element could not be named after a living person, even though the IUPAC had given the LBNL team the sole credit for its discovery. In 1997, IUPAC renamed elements 104 to 109, and gave element 104 the current name rutherfordium. The name dubnium was given to element 105 at the same time.
Isotopes
| Isotope |
Half-life |
Decay mode |
Discovery year |
Reaction |
|---|---|---|---|---|
| 253Rf | 48 μs | α, SF | 1994 | 204Pb(50Ti,n) |
| 254Rf | 23 μs | SF | 1994 | 206Pb(50Ti,2n) |
| 255Rf | 2.3 s | ε?, α, SF | 1974 | 207Pb(50Ti,2n) |
| 256Rf | 6.4 ms | α, SF | 1974 | 208Pb(50Ti,2n) |
| 257Rf | 4.7 s | ε, α, SF | 1969 | 249Cf(12C,4n) |
| 257mRf | 4.1 s | ε, α, SF | 1969 | 249Cf(12C,4n) |
| 258Rf | 14.7 ms | α, SF | 1969 | 249Cf(13C,4n) |
| 259Rf | 3.2 s | α, SF | 1969 | 249Cf(13C,3n) |
| 259mRf | 2.5 s | ε | 1969 | 249Cf(13C,3n) |
| 260Rf | 21 ms | α, SF | 1969 | 248Cm(16O,4n) |
| 261Rf | 78 s | α, SF | 1970 | 248Cm(18O,5n) |
| 261mRf | 4 s | ε, α, SF | 2001 | 244Pu(22Ne,5n) |
| 262Rf | 2.3 s | α, SF | 1996 | 244Pu(22Ne,4n) |
| 263Rf | 15 min | α, SF | 1999 |
263Db( e, ν e) |
| 263mRf ? | 8 s | α, SF | 1999 |
263Db( e, ν e) |
| 265Rf | 1.1 min | SF | 2010 | 269Sg(—,α) |
| 266Rf | 23 s? | SF | 2007? |
266Db( e, ν e)? |
| 267Rf | 48 min | SF | 2004 | 271Sg(—,α) |
| 268Rf | 1.4 s? | SF | 2004? |
268Db( e, ν e)? |
| 270Rf | 20 ms? | SF | 2010? |
270Db( e, ν e)? |
Rutherfordium has no stable or naturally occurring isotopes. Several radioactive isotopes have been synthesized in the laboratory, either by fusing two atoms or by observing the decay of heavier elements. Sixteen different isotopes have been reported with atomic masses from 253 to 270 (with the exceptions of 264 and 269). Most of these decay predominantly through spontaneous fission pathways.
Stability and half-lives
Out of isotopes whose half-lives are known, the lighter isotopes usually have shorter half-lives; half-lives of under 50 μs for 253Rf and 254Rf were observed. 256Rf, 258Rf, 260Rf are more stable at around 10 ms, 255Rf, 257Rf, 259Rf, and 262Rf live between 1 and 5 seconds, and 261Rf, 265Rf, and 263Rf are more stable, at around 1.1, 1.5, and 10 minutes respectively. The heaviest isotopes are the most stable, with 267Rf having a measured half-life of about 48 minutes.
The lightest isotopes were synthesized by direct fusion between two lighter nuclei and as decay products. The heaviest isotope produced by direct fusion is 262Rf; heavier isotopes have only been observed as decay products of elements with larger atomic numbers. The heavy isotopes 266Rf and 268Rf have also been reported as electron capture daughters of the dubnium isotopes 266Db and 268Db, but have short half-lives to spontaneous fission. It seems likely that the same is true for 270Rf, a possible daughter of 270Db. These three isotopes remain unconfirmed.
In 1999, American scientists at the University of California, Berkeley, announced that they had succeeded in synthesizing three atoms of 293Og. These parent nuclei were reported to have successively emitted seven alpha particles to form 265Rf nuclei, but their claim was retracted in 2001. This isotope was later discovered in 2010 as the final product in the decay chain of 285Fl.
Predicted properties
Very few properties of rutherfordium or its compounds have been measured; this is due to its extremely limited and expensive production and the fact that rutherfordium (and its parents) decays very quickly. A few singular chemistry-related properties have been measured, but properties of rutherfordium metal remain unknown and only predictions are available.
Chemical
Rutherfordium is the first transactinide element and the second member of the 6d series of transition metals. Calculations on its ionization potentials, atomic radius, as well as radii, orbital energies, and ground levels of its ionized states are similar to that of hafnium and very different from that of lead. Therefore, it was concluded that rutherfordium's basic properties will resemble those of other group 4 elements, below titanium, zirconium, and hafnium. Some of its properties were determined by gas-phase experiments and aqueous chemistry. The oxidation state +4 is the only stable state for the latter two elements and therefore rutherfordium should also exhibit a stable +4 state. In addition, rutherfordium is also expected to be able to form a less stable +3 state. The standard reduction potential of the Rf4+/Rf couple is predicted to be higher than −1.7 V.
Initial predictions of the chemical properties of rutherfordium were based on calculations which indicated that the relativistic effects on the electron shell might be strong enough that the 7p orbitals would have a lower energy level than the 6d orbitals, giving it a valence electron configuration of 6d1 7s2 7p1 or even 7s2 7p2, therefore making the element behave more like lead than hafnium. With better calculation methods and experimental studies of the chemical properties of rutherfordium compounds it could be shown that this does not happen and that rutherfordium instead behaves like the rest of the group 4 elements. Later it was shown in ab initio calculations with the high level of accuracy that the Rf atom has the ground state with the 6d2 7s2 valence configuration and the low-lying excited 6d1 7s2 7p1 state with the excitation energy of only 0.3–0.5 eV.
In an analogous manner to zirconium and hafnium, rutherfordium is projected to form a very stable, refractory oxide, RfO2. It reacts with halogens to form tetrahalides, RfX4, which hydrolyze on contact with water to form oxyhalides RfOX2. The tetrahalides are volatile solids existing as monomeric tetrahedral molecules in the vapor phase.
In the aqueous phase, the Rf4+ ion hydrolyzes less than titanium(IV) and to a similar extent as zirconium and hafnium, thus resulting in the RfO2+ ion. Treatment of the halides with halide ions promotes the formation of complex ions. The use of chloride and bromide ions produces the hexahalide complexes RfCl2−
6 and RfBr2−
6. For the fluoride complexes, zirconium and hafnium tend to form hepta- and octa- complexes. Thus, for the larger rutherfordium ion, the complexes RfF2−
6, RfF3−
7 and RfF4−
8 are possible.
Physical and atomic
Rutherfordium is expected to be a solid under normal conditions and have a hexagonal close-packed crystal structure (c/a = 1.61), similar to its lighter congener hafnium. It should be a metal with density ~17 g/cm3. The atomic radius of rutherfordium is expected to be ~150 pm. Due to relativistic stabilization of the 7s orbital and destabilization of the 6d orbital, Rf+ and Rf2+ ions are predicted to give up 6d electrons instead of 7s electrons, which is the opposite of the behavior of its lighter homologs. When under high pressure (variously calculated as 72 or ~50 GPa), rutherfordium is expected to transition to body-centered cubic crystal structure; hafnium transforms to this structure at 71±1 GPa, but has an intermediate ω structure that it transforms to at 38±8 GPa that should be lacking for rutherfordium.
Experimental chemistry
Gas phase

Early work on the study of the chemistry of rutherfordium focused on gas thermochromatography and measurement of relative deposition temperature adsorption curves. The initial work was carried out at Dubna in an attempt to reaffirm their discovery of the element. Recent work is more reliable regarding the identification of the parent rutherfordium radioisotopes. The isotope 261mRf has been used for these studies, though the long-lived isotope 267Rf (produced in the decay chain of 291Lv, 287Fl, and 283Cn) may be advantageous for future experiments. The experiments relied on the expectation that rutherfordium would be a 6d element in group 4 and should therefore form a volatile molecular tetrachloride, that would be tetrahedral in shape. Rutherfordium(IV) chloride is more volatile than its lighter homologue hafnium(IV) chloride (HfCl4) because its bonds are more covalent.
A series of experiments confirmed that rutherfordium behaves as a typical member of group 4, forming a tetravalent chloride (RfCl4) and bromide (RfBr4) as well as an oxychloride (RfOCl2). A decreased volatility was observed for RfCl
4 when potassium chloride is provided as the solid phase instead of gas, highly indicative of the formation of nonvolatile K
2RfCl
6 mixed salt.
Aqueous phase
Rutherfordium is expected to have the electron configuration [Rn]5f14 6d2 7s2 and therefore behave as the heavier homologue of hafnium in group 4 of the periodic table. It should therefore readily form a hydrated Rf4+ ion in strong acid solution and should readily form complexes in hydrochloric acid, hydrobromic or hydrofluoric acid solutions.
The most conclusive aqueous chemistry studies of rutherfordium have been performed by the Japanese team at Japan Atomic Energy Research Institute using the isotope 261mRf. Extraction experiments from hydrochloric acid solutions using isotopes of rutherfordium, hafnium, zirconium, as well as the pseudo-group 4 element thorium have proved a non-actinide behavior for rutherfordium. A comparison with its lighter homologues placed rutherfordium firmly in group 4 and indicated the formation of a hexachlororutherfordate complex in chloride solutions, in a manner similar to hafnium and zirconium.
-
261m
Rf4+
+ 6 Cl−
→ [261mRfCl
6]2−
Very similar results were observed in hydrofluoric acid solutions. Differences in the extraction curves were interpreted as a weaker affinity for fluoride ion and the formation of the hexafluororutherfordate ion, whereas hafnium and zirconium ions complex seven or eight fluoride ions at the concentrations used:
-
261m
Rf4+
+ 6 F−
→ [261mRfF
6]2−
Experiments performed in mixed sulfuric and nitric acid solutions shows that rutherfordium has a much weaker affinity towards forming sulfate complexes than hafnium. This result is in agreement with predictions, which expect rutherfordium complexes to be less stable than those of zirconium and hafnium because of a smaller ionic contribution to the bonding. This arises because rutherfordium has a larger ionic radius (76 pm) than zirconium (71 pm) and hafnium (72 pm), and also because of relativistic stabilisation of the 7s orbital and destabilisation and spin–orbit splitting of the 6d orbitals.
Coprecipitation experiments performed in 2021 studied rutherfordium's behaviour in basic solution containing ammonia or sodium hydroxide, using zirconium, hafnium, and thorium as comparisons. It was found that rutherfordium does not strongly coordinate with ammonia and instead coprecipitates out as a hydroxide, which is probably Rf(OH)4.