Lawrencium
| Lawrencium | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pronunciation |
|
||||||||||||||||||||||||||||||||||||||||||||
| Appearance | silvery (predicted) | ||||||||||||||||||||||||||||||||||||||||||||
| Mass number | [266] | ||||||||||||||||||||||||||||||||||||||||||||
| Lawrencium in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 103 | ||||||||||||||||||||||||||||||||||||||||||||
| Group | group 3 | ||||||||||||||||||||||||||||||||||||||||||||
| Period | period 7 | ||||||||||||||||||||||||||||||||||||||||||||
| Block | d-block | ||||||||||||||||||||||||||||||||||||||||||||
| Electron configuration | [Rn] 5f14 7s2 7p1 | ||||||||||||||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 8, 18, 32, 32, 8, 3 | ||||||||||||||||||||||||||||||||||||||||||||
| Physical properties | |||||||||||||||||||||||||||||||||||||||||||||
| Phase at STP | solid (predicted) | ||||||||||||||||||||||||||||||||||||||||||||
| Melting point | 1900 K (1600 °C, 3000 °F) (predicted) | ||||||||||||||||||||||||||||||||||||||||||||
| Density (near r.t.) | 14.4 g/cm3 (predicted) | ||||||||||||||||||||||||||||||||||||||||||||
| Atomic properties | |||||||||||||||||||||||||||||||||||||||||||||
| Oxidation states | +3 | ||||||||||||||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 1.3 (predicted) | ||||||||||||||||||||||||||||||||||||||||||||
| Ionization energies |
|
||||||||||||||||||||||||||||||||||||||||||||
| Other properties | |||||||||||||||||||||||||||||||||||||||||||||
| Natural occurrence | synthetic | ||||||||||||||||||||||||||||||||||||||||||||
| Crystal structure | hexagonal close-packed (hcp) (predicted) |
||||||||||||||||||||||||||||||||||||||||||||
| CAS Number | 22537-19-5 | ||||||||||||||||||||||||||||||||||||||||||||
| History | |||||||||||||||||||||||||||||||||||||||||||||
| Naming | after Ernest Lawrence | ||||||||||||||||||||||||||||||||||||||||||||
| Discovery | Lawrence Berkeley National Laboratory and Joint Institute for Nuclear Research (1961–1971) | ||||||||||||||||||||||||||||||||||||||||||||
| Isotopes of lawrencium | |||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||
Lawrencium is a synthetic chemical element; it has symbol Lr (formerly Lw) and atomic number 103. It is named in honor of Ernest Lawrence, inventor of the cyclotron, a device that was used to discover many artificial radioactive elements. A radioactive metal, lawrencium is the eleventh transuranic element and the last member of the actinide series. Like all elements with atomic number over 100, lawrencium can only be produced in particle accelerators by bombarding lighter elements with charged particles. Fourteen isotopes of lawrencium are currently known; the most stable is 266Lr with half-life 11 hours, but the shorter-lived 260Lr (half-life 2.7 minutes) is most commonly used in chemistry because it can be produced on a larger scale.
Chemistry experiments confirm that lawrencium behaves as a heavier homolog to lutetium in the periodic table, and is a trivalent element. It thus could also be classified as the first of the 7th-period transition metals: however, its electron configuration is anomalous for its position in the periodic table, having an s2p configuration instead of the s2d configuration of its homolog lutetium. This means that lawrencium may be more volatile than expected for its position in the periodic table and have a volatility comparable to that of lead.
In the 1950s, 1960s, and 1970s, many claims of the synthesis of lawrencium of varying quality were made from laboratories in the Soviet Union and the United States. The priority of the discovery and therefore the name of the element was disputed between Soviet and American scientists, and while the International Union of Pure and Applied Chemistry (IUPAC) initially established lawrencium as the official name for the element and gave the American team credit for the discovery, this was reevaluated in 1997, giving both teams shared credit for the discovery but not changing the element's name.
Introduction
Synthesis of superheavy nuclei
A superheavy atomic nucleus is created in a nuclear reaction that combines two other nuclei of unequal size into one; roughly, the more unequal the two nuclei in terms of mass, the greater the possibility that the two react. The material made of the heavier nuclei is made into a target, which is then bombarded by the beam of lighter nuclei. Two nuclei can only fuse into one if they approach each other closely enough; normally, nuclei (all positively charged) repel each other due to electrostatic repulsion. The strong interaction can overcome this repulsion but only within a very short distance from a nucleus; beam nuclei are thus greatly accelerated in order to make such repulsion insignificant compared to the velocity of the beam nucleus. The energy applied to the beam nuclei to accelerate them can cause them to reach speeds as high as one-tenth of the speed of light. However, if too much energy is applied, the beam nucleus can fall apart.
Coming close enough alone is not enough for two nuclei to fuse: when two nuclei approach each other, they usually remain together for approximately 10−20 seconds and then part ways (not necessarily in the same composition as before the reaction) rather than form a single nucleus. This happens because during the attempted formation of a single nucleus, electrostatic repulsion tears apart the nucleus that is being formed. Each pair of a target and a beam is characterized by its cross section—the probability that fusion will occur if two nuclei approach one another expressed in terms of the transverse area that the incident particle must hit in order for the fusion to occur. This fusion may occur as a result of the quantum effect in which nuclei can tunnel through electrostatic repulsion. If the two nuclei can stay close for past that phase, multiple nuclear interactions result in redistribution of energy and an energy equilibrium.
| External videos | |
|---|---|
|
|
The resulting merger is an excited state—termed a compound nucleus—and thus it is very unstable. To reach a more stable state, the temporary merger may fission without formation of a more stable nucleus. Alternatively, the compound nucleus may eject a few neutrons, which would carry away the excitation energy; if the latter is not sufficient for a neutron expulsion, the merger would produce a gamma ray. This happens in approximately 10−16 seconds after the initial nuclear collision and results in creation of a more stable nucleus. The definition by the IUPAC/IUPAP Joint Working Party (JWP) states that a chemical element can only be recognized as discovered if a nucleus of it has not decayed within 10−14 seconds. This value was chosen as an estimate of how long it takes a nucleus to acquire its outer electrons and thus display its chemical properties.
Decay and detection
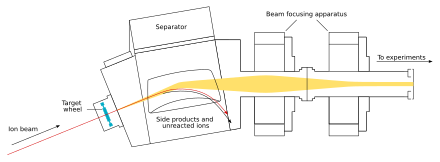
The beam passes through the target and reaches the next chamber, the separator; if a new nucleus is produced, it is carried with this beam. In the separator, the newly produced nucleus is separated from other nuclides (that of the original beam and any other reaction products) and transferred to a surface-barrier detector, which stops the nucleus. The exact location of the upcoming impact on the detector is marked; also marked are its energy and the time of the arrival. The transfer takes about 10−6 seconds; in order to be detected, the nucleus must survive this long. The nucleus is recorded again once its decay is registered, and the location, the energy, and the time of the decay are measured.
Stability of a nucleus is provided by the strong interaction. However, its range is very short; as nuclei become larger, its influence on the outermost nucleons (protons and neutrons) weakens. At the same time, the nucleus is torn apart by electrostatic repulsion between protons, and its range is not limited. Total binding energy provided by the strong interaction increases linearly with the number of nucleons, whereas electrostatic repulsion increases with the square of the atomic number, i.e. the latter grows faster and becomes increasingly important for heavy and superheavy nuclei. Superheavy nuclei are thus theoretically predicted and have so far been observed to predominantly decay via decay modes that are caused by such repulsion: alpha decay and spontaneous fission. Almost all alpha emitters have over 210 nucleons, and the lightest nuclide primarily undergoing spontaneous fission has 238. In both decay modes, nuclei are inhibited from decaying by corresponding energy barriers for each mode, but they can be tunnelled through.
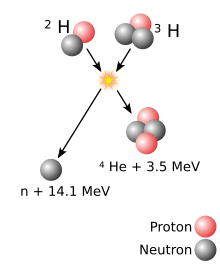
Alpha particles are commonly produced in radioactive decays because mass of an alpha particle per nucleon is small enough to leave some energy for the alpha particle to be used as kinetic energy to leave the nucleus. Spontaneous fission is caused by electrostatic repulsion tearing the nucleus apart and produces various nuclei in different instances of identical nuclei fissioning. As the atomic number increases, spontaneous fission rapidly becomes more important: spontaneous fission partial half-lives decrease by 23 orders of magnitude from uranium (element 92) to nobelium (element 102), and by 30 orders of magnitude from thorium (element 90) to fermium (element 100). The earlier liquid drop model thus suggested that spontaneous fission would occur nearly instantly due to disappearance of the fission barrier for nuclei with about 280 nucleons. The later nuclear shell model suggested that nuclei with about 300 nucleons would form an island of stability in which nuclei will be more resistant to spontaneous fission and will primarily undergo alpha decay with longer half-lives. Subsequent discoveries suggested that the predicted island might be further than originally anticipated; they also showed that nuclei intermediate between the long-lived actinides and the predicted island are deformed, and gain additional stability from shell effects. Experiments on lighter superheavy nuclei, as well as those closer to the expected island, have shown greater than previously anticipated stability against spontaneous fission, showing the importance of shell effects on nuclei.
Alpha decays are registered by the emitted alpha particles, and the decay products are easy to determine before the actual decay; if such a decay or a series of consecutive decays produces a known nucleus, the original product of a reaction can be easily determined. (That all decays within a decay chain were indeed related to each other is established by the location of these decays, which must be in the same place.) The known nucleus can be recognized by the specific characteristics of decay it undergoes such as decay energy (or more specifically, the kinetic energy of the emitted particle). Spontaneous fission, however, produces various nuclei as products, so the original nuclide cannot be determined from its daughters.
The information available to physicists aiming to synthesize a superheavy element is thus the information collected at the detectors: location, energy, and time of arrival of a particle to the detector, and those of its decay. The physicists analyze this data and seek to conclude that it was indeed caused by a new element and could not have been caused by a different nuclide than the one claimed. Often, provided data is insufficient for a conclusion that a new element was definitely created and there is no other explanation for the observed effects; errors in interpreting data have been made.History

In 1958, scientists at Lawrence Berkeley National Laboratory claimed the discovery of element 102, now called nobelium. At the same time, they also tried to synthesize element 103 by bombarding the same curium target used with nitrogen-14 ions. Eighteen tracks were noted, with decay energy around 9±1 MeV and half-life around 0.25 s; the Berkeley team noted that while the cause could be the production of an isotope of element 103, other possibilities could not be ruled out. While the data agrees reasonably with that later discovered for 257Lr (alpha decay energy 8.87 MeV, half-life 0.6 s), the evidence obtained in this experiment fell far short of the strength required to conclusively demonstrate synthesis of element 103. A follow-up on this experiment was not done, as the target was destroyed. Later, in 1960, the Lawrence Berkeley Laboratory attempted to synthesize the element by bombarding 252Cf with 10B and 11B. The results of this experiment were not conclusive.
The first important work on element 103 was done at Berkeley by the nuclear-physics team of Albert Ghiorso, Torbjørn Sikkeland, Almon Larsh, Robert M. Latimer, and their co-workers on February 14, 1961. The first atoms of lawrencium were reportedly made by bombarding a three-milligram target consisting of three isotopes of californium with boron-10 and boron-11 nuclei from the Heavy Ion Linear Accelerator (HILAC). The Berkeley team reported that the isotope 257103 was detected in this manner, and that it decayed by emitting an 8.6 MeV alpha particle with a half-life of 8±2 s. This identification was later corrected to 258103, as later work proved that 257Lr did not have the properties detected, but 258Lr did. This was considered at the time to be convincing proof of synthesis of element 103: while the mass assignment was less certain and proved to be mistaken, it did not affect the arguments in favor of element 103 having been synthesized. Scientists at Joint Institute for Nuclear Research in Dubna (then in the Soviet Union) raised several criticisms: all but one were answered adequately. The exception was that 252Cf was the most common isotope in the target, and in the reactions with 10B, 258Lr could only have been produced by emitting four neutrons, and emitting three neutrons was expected to be much less likely than emitting four or five. This would lead to a narrow yield curve, not the broad one reported by the Berkeley team. A possible explanation was that there was a low number of events attributed to element 103. This was an important intermediate step to the unquestioned discovery of element 103, although the evidence was not completely convincing. The Berkeley team proposed the name "lawrencium" with symbol "Lw", after Ernest Lawrence, inventor of the cyclotron. The IUPAC Commission on Nomenclature of Inorganic Chemistry accepted the name, but changed the symbol to "Lr". This acceptance of the discovery was later characterized as being hasty by the Dubna team.
-
252
98Cf + 11
5B → 263
103Lr* → 258
103Lr + 5 1
0n
The first work at Dubna on element 103 came in 1965, when they reported to have made 256103 in 1965 by bombarding 243Am with 18O, identifying it indirectly from its granddaughter fermium-252. The half-life they reported was somewhat too high, possibly due to background events. Later 1967 work on the same reaction identified two decay energies in the ranges 8.35–8.50 MeV and 8.50–8.60 MeV: these were assigned to 256103 and 257103. Despite repeat attempts, they were unable to confirm assignment of an alpha emitter with a half-life of 8 seconds to 257103. The Russians proposed the name "rutherfordium" for the new element in 1967: this name was later proposed by Berkeley for element 104.
-
243
95Am + 18
8O → 261
103Lr* → 256
103Lr + 5 1
0n
Further experiments in 1969 at Dubna and in 1970 at Berkeley demonstrated an actinide chemistry for the new element; so by 1970 it was known that element 103 is the last actinide. In 1970, the Dubna group reported the synthesis of 255103 with half-life 20 s and alpha decay energy 8.38 MeV. However, it was not until 1971, when the nuclear physics team at University of California at Berkeley successfully did a whole series of experiments aimed at measuring the nuclear decay properties of the lawrencium isotopes with mass numbers 255 to 260, that all previous results from Berkeley and Dubna were confirmed, apart from the Berkeley's group initial erroneous assignment of their first produced isotope to 257103 instead of the probably correct 258103. All final doubts were dispelled in 1976 and 1977 when the energies of X-rays emitted from 258103 were measured.

In 1971, the IUPAC granted the discovery of lawrencium to the Lawrence Berkeley Laboratory, even though they did not have ideal data for the element's existence. But in 1992, the IUPAC Transfermium Working Group (TWG) officially recognized the nuclear physics teams at Dubna and Berkeley as co-discoverers of lawrencium, concluding that while the 1961 Berkeley experiments were an important step to lawrencium's discovery, they were not yet fully convincing; and while the 1965, 1968, and 1970 Dubna experiments came very close to the needed level of confidence taken together, only the 1971 Berkeley experiments, which clarified and confirmed previous observations, finally resulted in complete confidence in the discovery of element 103. Because the name "lawrencium" had been in use for a long time by this point, it was retained by IUPAC, and in August 1997, the International Union of Pure and Applied Chemistry (IUPAC) ratified the name lawrencium and the symbol "Lr" during a meeting in Geneva.
Characteristics
Physical
Lawrencium is the last actinide. Authors considering the subject generally consider it a group 3 element, along with scandium, yttrium, and lutetium, as its filled f-shell is expected to make it resemble the other 7th-period transition metals. In the periodic table, it is to the right of the actinide nobelium, to the left of the 6d transition metal rutherfordium, and under the lanthanide lutetium with which it shares many physical and chemical properties. Lawrencium is expected to be a solid under normal conditions and have a hexagonal close-packed crystal structure (c/a = 1.58), similar to its lighter congener lutetium, though this is not yet known experimentally. The enthalpy of sublimation of lawrencium is estimated at 352 kJ/mol, close to the value of lutetium and strongly suggesting that metallic lawrencium is trivalent with three electrons delocalized, a prediction also supported by a systematic extrapolation of the values of heat of vaporization, bulk modulus, and atomic volume of neighboring elements to lawrencium: this makes it unlike the immediately preceding late actinides which are known to be (fermium and mendelevium) or expected to be (nobelium) divalent. The estimated enthalpies of vaporization show that lawrencium deviates from the trend of the late actinides and instead matches the trend of the succeeding 6d elements rutherfordium and dubnium, consistent with lawrencium's interpretation as a group 3 element. Some scientists prefer to end the actinides with nobelium and consider lawrencium to be the first transition metal of the seventh period.
Specifically, lawrencium is expected to be a trivalent, silvery metal, easily oxidized by air, steam, and acids, and having an atomic volume similar to that of lutetium and a trivalent metallic radius of 171 pm. It is expected to be a rather heavy metal with a density of around 14.4 g/cm3. It is also predicted to have a melting point of around 1900 K (1600 °C), not far from the value for lutetium (1925 K).
Chemical

In 1949, Glenn T. Seaborg, who devised the actinide concept, predicted that element 103 (lawrencium) should be the last actinide and that the Lr3+ ion should be about as stable as Lu3+ in aqueous solution. It was not until decades later that element 103 was finally conclusively synthesized and this prediction was experimentally confirmed.
1969 studies on the element showed that lawrencium reacts with chlorine to form a product that was most likely the trichloride, LrCl3. Its volatility was found to be similar to the chlorides of curium, fermium, and nobelium and much less than that of rutherfordium chloride. In 1970, chemical studies were performed on 1500 atoms of 256Lr, comparing it with divalent (No, Ba, Ra), trivalent (Fm, Cf, Cm, Am, Ac), and tetravalent (Th, Pu) elements. It was found that lawrencium coextracted with the trivalent ions, but the short half-life of 256Lr precluded a confirmation that it eluted ahead of Md3+ in the elution sequence. Lawrencium occurs as the trivalent Lr3+ ion in aqueous solution and hence its compounds should be similar to those of the other trivalent actinides: for example, lawrencium(III) fluoride (LrF3) and hydroxide (Lr(OH)3) should both be insoluble in water. Due to the actinide contraction, the ionic radius of Lr3+ should be smaller than that of Md3+, and it should elute ahead of Md3+ when ammonium α-hydroxyisobutyrate (ammonium α-HIB) is used as an eluant. Later 1987 experiments on the longer-lived isotope 260Lr confirmed lawrencium's trivalency and that it eluted in roughly the same place as erbium, and found that lawrencium's ionic radius was 88.6±0.3 pm, larger than would be expected from simple extrapolation from periodic trends. Later 1988 experiments with more lawrencium atoms refined this to 88.1±0.1 pm and calculated an enthalpy of hydration value of −3685±13 kJ/mol. It was also found that the actinide contraction at the end of the actinides was larger than the analogous lanthanide contraction, with the exception of the last actinide, lawrencium: the cause was speculated to be relativistic effects.
It has been speculated that the 7s electrons are relativistically stabilized, so that in reducing conditions, only the 7p1/2 electron would be ionized, leading to the monovalent Lr+ ion. However, all experiments to reduce Lr3+ to Lr2+ or Lr+ in aqueous solution were unsuccessful, similarly to lutetium. On the basis of this, the standard electrode potential of the E°(Lr3+ → Lr+) couple was calculated to be less than −1.56 V, indicating that the existence of Lr+ ions in aqueous solution was unlikely. The upper limit for the E°(Lr3+ → Lr2+) couple was predicted to be −0.44 V: the values for E°(Lr3+ → Lr) and E°(Lr4+ → Lr3+) are predicted to be −2.06 V and +7.9 V. The stability of the group oxidation state in the 6d transition series decreases as RfIV > DbV > SgVI, and lawrencium continues the trend with LrIII being more stable than RfIV.
In the molecule lawrencium dihydride (LrH2), which is predicted to be bent, the 6d orbital of lawrencium is not expected to play a role in the bonding, unlike that of lanthanum dihydride (LaH2). LaH2 has La–H bond distances of 2.158 Å, while LrH2 should have shorter Lr–H bond distances of 2.042 Å due to the relativistic contraction and stabilization of the 7s and 7p orbitals involved in the bonding, in contrast to the core-like 5f subshell and the mostly uninvolved 6d subshell. In general, molecular LrH2 and LrH are expected to resemble the corresponding thallium species (thallium having a 6s26p1 valence configuration in the gas phase, like lawrencium's 7s27p1) more than the corresponding lanthanide species. The electron configurations of Lr+ and Lr2+ are expected to be 7s2 and 7s1 respectively. However, in species where all three valence electrons of lawrencium are ionized to give at least formally the Lr3+ cation, lawrencium is expected to behave like a typical actinide and the heavier congener of lutetium, especially because the first three ionization potentials of lawrencium are predicted to be similar to those of lutetium. Hence, unlike thallium but like lutetium, lawrencium would prefer to form LrH3 than LrH, and LrCO is expected to be similar to the also unknown LuCO, both metals having valence configuration σ2π1 in their monocarbonyls. The pπ–dπ bond is expected to be seen in LrCl3 just as it is for LuCl3 and more generally all the LnCl3. The complex anion [Lr(C5H4SiMe3)3]− is expected to be stable with a configuration of 6d1 for lawrencium; this 6d orbital would be its highest occupied molecular orbital. This is analogous to the electronic structure of the analogous lutetium compound.
Atomic
Lawrencium has three valence electrons: the 5f electrons are in the atomic core. In 1970, it was predicted that the ground-state electron configuration of lawrencium was [Rn]5f146d17s2 (ground state term symbol 2D3/2), per the Aufbau principle and conforming to the [Xe]4f145d16s2 configuration of lawrencium's lighter homolog lutetium. But the next year, calculations were published that questioned this prediction, instead expecting an anomalous [Rn]5f147s27p1 configuration. Though early calculations gave conflicting results, more recent studies and calculations confirm the s2p suggestion. 1974 relativistic calculations concluded that the energy difference between the two configurations was small and that it was uncertain which was the ground state. Later 1995 calculations concluded that the s2p configuration should be energetically favored, because the spherical s and p1/2 orbitals are nearest to the atomic nucleus and thus move quickly enough that their relativistic mass increases significantly.
In 1988, a team of scientists led by Eichler calculated that lawrencium's enthalpy of adsorption on metal sources would differ enough depending on its electron configuration that it would be feasible to carry out experiments to exploit this fact to measure lawrencium's electron configuration. The s2p configuration was expected to be more volatile than the s2d configuration, and be more similar to that of the p-block element lead. No evidence for lawrencium being volatile was obtained and the lower limit for the enthalpy of adsorption of lawrencium on quartz or platinum was significantly higher than the estimated value for the s2p configuration.

In 2015, the first ionization energy of lawrencium was measured, using the isotope 256Lr. The measured value, 4.96+0.08
−0.07 eV, agreed very well with the relativistic theoretical prediction of 4.963(15) eV, and also provided a first step into measuring the first ionization energies of the transactinides. This value is the lowest among all the lanthanides and actinides, and supports the s2p configuration as the 7p1/2 electron is expected to be only weakly bound. As ionisation energies generally increase left to right in the f-block, this low value suggests that lutetium and lawrencium belong in the d-block (whose trend they follow) and not the f-block. That would make them the heavier congeners of scandium and yttrium, rather than lanthanum and actinium. Although some alkali metal-like behaviour has been predicted, adsorption experiments suggest that lawrencium is trivalent like scandium and yttrium, not monovalent like the alkali metals. A lower limit on lawrencium's second ionization energy (>13.3 eV) was experimentally found in 2021.
Even though s2p is now known to be the ground-state configuration of the lawrencium atom, ds2 should be a low-lying excited-state configuration, with an excitation energy variously calculated as 0.156 eV, 0.165 eV, or 0.626 eV. As such lawrencium may still be considered to be a d-block element, albeit with an anomalous electron configuration (like chromium or copper), as its chemical behaviour matches expectations for a heavier analogue of lutetium.
Isotopes
Fourteen isotopes of lawrencium are known, with mass number 251–262, 264, and 266; all are radioactive. Seven nuclear isomers are known. The longest-lived isotope, 266Lr, has a half-life of about ten hours and is one of the longest-lived superheavy isotopes known to date. However, shorter-lived isotopes are usually used in chemical experiments because 266Lr currently can only be produced as a final decay product of even heavier and harder-to-make elements: it was discovered in 2014 in the decay chain of 294Ts. 256Lr (half-life 27 seconds) was used in the first chemical studies on lawrencium: currently, the longer-lived 260Lr (half-life 2.7 minutes) is usually used for this purpose. After 266Lr, the longest-lived isotopes are 264Lr (4.8+2.2
−1.3 h), 262Lr (3.6 h), and 261Lr (44 min). All other known lawrencium isotopes have half-lives under 5 minutes, and the shortest-lived of them (251Lr) has a half-life of 24.4 milliseconds. The half-lives of lawrencium isotopes mostly increase smoothly from 251Lr to 266Lr, with a dip from 257Lr to 259Lr.
Preparation and purification
Most isotopes of lawrencium can be produced by bombarding actinide (americium to einsteinium) targets with light ions (from boron to neon). The two most important isotopes, 256Lr and 260Lr, can be respectively produced by bombarding californium-249 with 70 MeV boron-11 ions (producing lawrencium-256 and four neutrons) and by bombarding berkelium-249 with oxygen-18 (producing lawrencium-260, an alpha particle, and three neutrons). The two heaviest and longest-lived known isotopes, 264Lr and 266Lr, can only be produced at much lower yields as decay products of dubnium, whose progenitors are isotopes of moscovium and tennessine.
Both 256Lr and 260Lr have half-lives too short to allow a complete chemical purification process. Early experiments with 256Lr therefore used rapid solvent extraction, with the chelating agent thenoyltrifluoroacetone (TTA) dissolved in methyl isobutyl ketone (MIBK) as the organic phase, and with the aqueous phase being buffered acetate solutions. Ions of different charge (+2, +3, or +4) will then extract into the organic phase under different pH ranges, but this method will not separate the trivalent actinides and thus 256Lr must be identified by its emitted 8.24 MeV alpha particles. More recent methods have allowed rapid selective elution with α-HIB to take place in enough time to separate out the longer-lived isotope 260Lr, which can be removed from the catcher foil with 0.05 M hydrochloric acid.